https://pmc.ncbi.nlm.nih.gov/articles/PMC4161296/
The Time Scale of Evolutionary Innovation
Resumo
Uma questão fundamental em biologia é a seguinte: qual é a escala de tempo necessária para inovações evolutivas? Existem muitos resultados que caracterizam etapas únicas em termos do tempo de fixação de novos mutantes surgindo em populações de certo tamanho e estrutura. Mas aqui fazemos uma pergunta diferente, que está relacionada à escala de tempo muito maior de trajetórias evolutivas: quanto tempo leva para uma população explorando uma paisagem de aptidão encontrar sequências-alvo que codifiquem novas funções biológicas? Nossa variável-chave é o comprimento 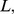 da sequência genética que sofre adaptação. Na ciência da computação, há uma distinção crucial entre problemas que requerem algoritmos que levam tempo polinomial ou exponencial. Os últimos são considerados intratáveis. Aqui, desenvolvemos uma abordagem teórica que nos permite estimar o tempo de evolução como função de
da sequência genética que sofre adaptação. Na ciência da computação, há uma distinção crucial entre problemas que requerem algoritmos que levam tempo polinomial ou exponencial. Os últimos são considerados intratáveis. Aqui, desenvolvemos uma abordagem teórica que nos permite estimar o tempo de evolução como função de 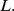 Mostramos que a adaptação em muitas paisagens de aptidão leva um tempo exponencial em
Mostramos que a adaptação em muitas paisagens de aptidão leva um tempo exponencial em 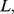 mesmo se houver amplos gradientes de seleção e muitos alvos uniformemente distribuídos no espaço de sequência. Esses resultados negativos nos levam a buscar mecanismos específicos que permitam que a evolução funcione em escalas de tempo polinomiais. Estudamos um processo de regeneração e mostramos que ele permite que a evolução funcione em tempo polinomial.
mesmo se houver amplos gradientes de seleção e muitos alvos uniformemente distribuídos no espaço de sequência. Esses resultados negativos nos levam a buscar mecanismos específicos que permitam que a evolução funcione em escalas de tempo polinomiais. Estudamos um processo de regeneração e mostramos que ele permite que a evolução funcione em tempo polinomial.
Resumo do autor
Adaptação evolutiva pode ser descrita como uma caminhada distorcida e estocástica de uma população de sequências em um espaço de sequência de alta dimensão. A população explora uma paisagem de aptidão. O processo de mutação-seleção distorce a população em direção a regiões de maior aptidão. Neste artigo, estimamos a escala de tempo necessária para a inovação evolutiva. Nosso parâmetro-chave é o comprimento da sequência genética que precisa ser adaptada. Mostramos que uma variedade de processos evolutivos leva tempo exponencial no comprimento da sequência. Propomos um processo específico, que chamamos de "processos de regeneração", e mostramos que ele permite que a evolução funcione em escalas de tempo polinomiais. Nessa visão, a evolução pode resolver um problema de forma eficiente se já tiver resolvido um problema semelhante.
Introdução
Nosso planeta surgiu há 4,6 bilhões de anos. Há evidências químicas claras de vida na Terra há 3,5 bilhões de anos [1] , [2] . O processo evolutivo gerou procárias, eucarias e organismos multicelulares complexos. Ao longo da história da vida, a evolução teve que descobrir sequências de polímeros biológicos que desempenham funções específicas e complicadas. O comprimento médio dos genes bacterianos é de cerca de 1.000 nucleotídeos, o dos genes humanos de cerca de 3.000 nucleotídeos. O maior gene bacteriano conhecido contém mais de 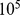 nucleotídeos, o maior gene humano mais de
nucleotídeos, o maior gene humano mais de 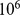 . Uma questão básica é qual é a escala de tempo necessária para a evolução descobrir as sequências que desempenham as funções desejadas. Embora existam muitos resultados para o tempo de fixação de mutantes individuais [3] – [15] , aqui perguntamos como a escala de tempo da evolução depende do comprimento
. Uma questão básica é qual é a escala de tempo necessária para a evolução descobrir as sequências que desempenham as funções desejadas. Embora existam muitos resultados para o tempo de fixação de mutantes individuais [3] – [15] , aqui perguntamos como a escala de tempo da evolução depende do comprimento 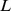 da sequência que precisa ser adaptada. Consideramos a distinção crucial entre tempo polinomial e exponencial [16] – [18] . Uma escala de tempo que cresce exponencialmente em
da sequência que precisa ser adaptada. Consideramos a distinção crucial entre tempo polinomial e exponencial [16] – [18] . Uma escala de tempo que cresce exponencialmente em 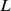 é inviável para sequências longas.
é inviável para sequências longas.
A dinâmica evolutiva opera no espaço de sequência, que pode ser imaginado como uma rede multidimensional discreta que surge quando todas as sequências de um determinado comprimento são organizadas de modo que os vizinhos mais próximos diferem por uma mutação pontual [19] . Para seleção constante, cada ponto no espaço de sequência é associado a um valor de aptidão não negativo (taxa reprodutiva). A paisagem de aptidão resultante é uma cadeia de montanhas de alta dimensão. As populações exploram paisagens de aptidão em busca de regiões elevadas, cristas e picos [20] – [27] .
Uma questão que tem sido extensivamente estudada é quanto tempo leva para que as funções biológicas existentes melhorem sob seleção natural. Esse problema leva ao estudo de caminhadas adaptativas em paisagens de aptidão [15] , [20] , [21] , [28] , [29] . Neste artigo, fazemos uma pergunta diferente: quanto tempo leva para a evolução descobrir uma nova função? Mais especificamente, nosso objetivo é estimar o tempo esperado de descoberta de novas funções biológicas: quanto tempo leva para uma população de organismos reprodutores descobrir uma função biológica que não está presente no início da busca. Discutiremos duas aproximações para paisagens de aptidão robustas. Também discutiremos a importância de picos agrupados.
Consideramos um alfabeto de tamanho quatro, como é o caso do DNA e do RNA, e uma sequência de nucleotídeos de comprimento 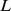 . Consideramos uma população de tamanho
. Consideramos uma população de tamanho 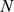 , que se reproduz assexuadamente. A taxa de mutação,
, que se reproduz assexuadamente. A taxa de mutação,  , é pequena: mutações individuais são introduzidas e avaliadas por seleção natural e deriva aleatória, uma de cada vez. A probabilidade de que o processo evolutivo se mova de uma sequência
, é pequena: mutações individuais são introduzidas e avaliadas por seleção natural e deriva aleatória, uma de cada vez. A probabilidade de que o processo evolutivo se mova de uma sequência  para uma sequência
para uma sequência 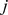 , que está à distância de Hamming um de
, que está à distância de Hamming um de  , é dada por
, é dada por 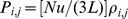 , onde
, onde 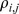 é a probabilidade de fixação de sequência
é a probabilidade de fixação de sequência 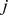 em uma população que consiste em sequência
em uma população que consiste em sequência  . No caso especial de uma paisagem de aptidão plana, temos
. No caso especial de uma paisagem de aptidão plana, temos 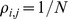 , e
, e 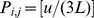 . Assim, temos uma caminhada aleatória evolutiva, onde cada passo é um salto para uma sequência vizinha de distância de Hamming um.
. Assim, temos uma caminhada aleatória evolutiva, onde cada passo é um salto para uma sequência vizinha de distância de Hamming um.
Resultados
Considere um espaço de sequência de alta dimensão. Uma função biológica específica pode ser instanciada por algumas das sequências. Cada sequência  tem um valor de aptidão
tem um valor de aptidão 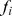 , que mede a capacidade da sequência
, que mede a capacidade da sequência  de codificar a função desejada. Normalmente, espera-se que paisagens de aptidão biológica tenham muitos picos [29] – [31] . Elas podem ser altamente robustas devido aos efeitos epistáticos de mutações [32] – [34] . Elas também podem conter grandes regiões ou redes de neutralidade [20] , [21] . Estudos empíricos de sequências curtas de RNA revelaram que a paisagem de aptidão subjacente tem baixa densidade de pico [35] : em torno de
de codificar a função desejada. Normalmente, espera-se que paisagens de aptidão biológica tenham muitos picos [29] – [31] . Elas podem ser altamente robustas devido aos efeitos epistáticos de mutações [32] – [34] . Elas também podem conter grandes regiões ou redes de neutralidade [20] , [21] . Estudos empíricos de sequências curtas de RNA revelaram que a paisagem de aptidão subjacente tem baixa densidade de pico [35] : em torno de 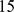 picos em
picos em 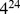 sequências.
sequências.
Para o propósito de estimar o tempo de descoberta esperado, podemos aproximar o cenário de aptidão com uma função de degrau binária sobre o espaço de sequência. Discutimos duas aproximações diferentes ( Figura 1 ). Para a primeira aproximação, consideramos o cenário em que valores de aptidão abaixo de algum limite, 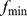 , têm contribuição insignificante; essas sequências não instanciam a função desejada (nem um pouco ou apenas abaixo do nível mínimo que poderia ser detectado pela seleção natural). Aproximamos o cenário de aptidão robusto da seguinte forma: se
, têm contribuição insignificante; essas sequências não instanciam a função desejada (nem um pouco ou apenas abaixo do nível mínimo que poderia ser detectado pela seleção natural). Aproximamos o cenário de aptidão robusto da seguinte forma: se 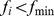 então
então 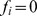 ; se
; se 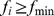 então
então 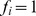 . O conjunto de sequências com
. O conjunto de sequências com 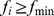 constitui o conjunto alvo, e o cenário de aptidão restante é neutro.
constitui o conjunto alvo, e o cenário de aptidão restante é neutro.
Figura 1. Aproximações de uma paisagem de aptidão altamente acidentada por picos amplos e regiões neutras.

As figuras descrevem exemplos de paisagens de aptidão altamente robustas onde o espaço de sequência foi projetado em uma dimensão. (A) Sequências com aptidão abaixo de algum nível 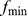 são funcionalmente muito diferentes da função desejada, e a seleção não pode atuar sobre elas. Todas as outras sequências são consideradas alvos. A paisagem de aptidão é aproximada por uma função degrau: se
são funcionalmente muito diferentes da função desejada, e a seleção não pode atuar sobre elas. Todas as outras sequências são consideradas alvos. A paisagem de aptidão é aproximada por uma função degrau: se 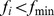 , então
, então 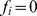 , caso contrário
, caso contrário 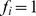 . (B) Máximos locais abaixo do limite de aptidão desejado
. (B) Máximos locais abaixo do limite de aptidão desejado 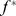 são conhecidos por desacelerar a caminhada aleatória evolutiva em direção a sequências que atingem aptidão pelo menos
são conhecidos por desacelerar a caminhada aleatória evolutiva em direção a sequências que atingem aptidão pelo menos 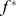 . Aproximamos a paisagem de aptidão por picos amplos e regiões neutras aumentando a aptidão de cada sequência que pertence a uma cadeia de montanhas com aptidão abaixo
. Aproximamos a paisagem de aptidão por picos amplos e regiões neutras aumentando a aptidão de cada sequência que pertence a uma cadeia de montanhas com aptidão abaixo 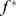 para os máximos locais máximos
para os máximos locais máximos 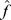 abaixo de
abaixo de 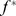 . Observe que o conjunto de alvos começa na encosta de uma cadeia de montanhas cujo pico excede
. Observe que o conjunto de alvos começa na encosta de uma cadeia de montanhas cujo pico excede 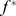 .
.
A segunda aproximação funciona da seguinte forma. Considere o processo evolutivo explorando uma paisagem de aptidão robusta onde o objetivo é atingir um nível de aptidão 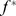 . Máximos locais abaixo
. Máximos locais abaixo 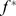 desaceleram o processo evolutivo para atingir
desaceleram o processo evolutivo para atingir 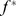 , porque a caminhada evolutiva pode ficar presa nesses máximos locais. Para derivar limites inferiores para o tempo de descoberta esperado, a paisagem de aptidão robusta pode ser aproximada da seguinte forma. Seja
, porque a caminhada evolutiva pode ficar presa nesses máximos locais. Para derivar limites inferiores para o tempo de descoberta esperado, a paisagem de aptidão robusta pode ser aproximada da seguinte forma. Seja 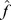 o valor de aptidão do máximo local mais alto abaixo
o valor de aptidão do máximo local mais alto abaixo 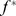 . Então, para cada sequência em uma cadeia de montanhas com um máximo local abaixo,
. Então, para cada sequência em uma cadeia de montanhas com um máximo local abaixo, 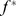 atribuímos o valor de aptidão
atribuímos o valor de aptidão 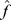 . As cadeias de montanhas com máximos locais acima
. As cadeias de montanhas com máximos locais acima 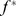 são as sequências alvo. Observe que o conjunto alvo inclui sequências que começam na encosta de cadeias de montanhas com picos acima
são as sequências alvo. Observe que o conjunto alvo inclui sequências que começam na encosta de cadeias de montanhas com picos acima 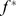 . Assim, novamente obtemos uma paisagem de aptidão com alvos agrupados e região neutra, onde a região neutra consiste em todas as sequências cujos valores de aptidão foram atribuídos a
. Assim, novamente obtemos uma paisagem de aptidão com alvos agrupados e região neutra, onde a região neutra consiste em todas as sequências cujos valores de aptidão foram atribuídos a 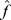 . As duas aproximações são ilustradas na Figura 1 . Pois
. As duas aproximações são ilustradas na Figura 1 . Pois 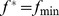 a segunda aproximação gera áreas alvo maiores do que a primeira aproximação e, portanto, é mais branda.
a segunda aproximação gera áreas alvo maiores do que a primeira aproximação e, portanto, é mais branda.
Nossos principais resultados para estimar o tempo de descoberta agora podem ser formulados para paisagens de aptidão binárias, mas eles se aplicam a qualquer tipo de paisagem acidentada usando uma das duas aproximações. Observamos que nossos métodos também podem ser aplicados para certas paisagens de aptidão não binárias, e um exemplo de uma paisagem de aptidão com um grande gradiente decorrente de efeitos de aptidão multiplicativos é discutido nas Seções 6 e 7 do Texto S1 .
Agora apresentamos nossos principais resultados na seguinte ordem. Primeiro estimamos o tempo de descoberta de uma única busca visando encontrar um único pico amplo. Então estudamos múltiplas buscas simultâneas para um único pico amplo. Finalmente, consideramos múltiplos picos amplos que são uniformemente distribuídos aleatoriamente no espaço de sequência.
Primeiro, estudamos um pico amplo de sequências alvo descritas da seguinte forma: considere uma sequência específica; qualquer sequência dentro de uma certa distância de Hamming dessa sequência pertence ao conjunto alvo. Especificamente, consideramos que o processo evolutivo foi bem-sucedido se a população descobrir uma sequência que difere da sequência específica em não mais do que uma fração  de posições. Nós nos referimos à sequência específica como o centro alvo e
de posições. Nós nos referimos à sequência específica como o centro alvo e  como a largura (ou raio) do pico. Por exemplo, se
como a largura (ou raio) do pico. Por exemplo, se 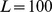 e
e 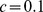 , então o centro alvo é cercado por uma nuvem de
, então o centro alvo é cercado por uma nuvem de 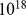 sequências aproximadamente. Para um único pico amplo com largura
sequências aproximadamente. Para um único pico amplo com largura  , o conjunto alvo contém pelo menos
, o conjunto alvo contém pelo menos 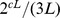 sequências, que é uma função exponencial de
sequências, que é uma função exponencial de 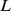 . O cenário de aptidão fora do pico amplo é plano. Nós nos referimos a esse cenário de aptidão binário como um cenário de pico amplo. A população precisa descobrir qualquer uma das sequências alvo no pico amplo, começando de alguma sequência que não esteja no pico amplo. Nós estabelecemos o seguinte resultado.
. O cenário de aptidão fora do pico amplo é plano. Nós nos referimos a esse cenário de aptidão binário como um cenário de pico amplo. A população precisa descobrir qualquer uma das sequências alvo no pico amplo, começando de alguma sequência que não esteja no pico amplo. Nós estabelecemos o seguinte resultado.
Teorema 1
Considere uma única busca explorando uma ampla paisagem de pico com largura e taxa de mutação . As seguintes afirmações são válidas :

se , então existe tal que para todos os espaços de sequência de comprimento de sequência , o tempo de descoberta esperado é pelo menos ;
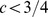
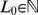
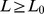
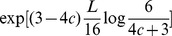
se , então para todos os espaços de sequência de comprimento de sequência , o tempo de descoberta esperado é no máximo .
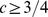
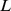
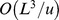
Nosso resultado pode ser interpretado da seguinte forma (veja Teorema S2 e Corolário S2 no Texto S1 ): (i) Se 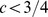 , então o tempo de descoberta esperado é exponencial em
, então o tempo de descoberta esperado é exponencial em 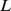 ; e (ii) se
; e (ii) se 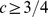 , então o tempo de descoberta esperado é polinomial em
, então o tempo de descoberta esperado é polinomial em 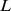 . Assim, derivamos um resultado de forte dicotomia que mostra uma transição brusca do tempo polinomial para o exponencial dependendo se uma condição específica em
. Assim, derivamos um resultado de forte dicotomia que mostra uma transição brusca do tempo polinomial para o exponencial dependendo se uma condição específica em  é ou não válida.
é ou não válida.
Para o alfabeto de quatro letras, a maioria das sequências aleatórias tem distância de Hamming 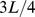 do centro alvo. Se a população estiver mais distante do que essa distância de Hamming, então a deriva aleatória a aproximará. Se a população estiver mais próxima do que essa distância de Hamming, então a deriva aleatória a afastará ainda mais. Esse argumento constitui a razão intuitiva que
do centro alvo. Se a população estiver mais distante do que essa distância de Hamming, então a deriva aleatória a aproximará. Se a população estiver mais próxima do que essa distância de Hamming, então a deriva aleatória a afastará ainda mais. Esse argumento constitui a razão intuitiva que 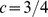 é o limite crítico. Se o pico tiver uma largura menor que
é o limite crítico. Se o pico tiver uma largura menor que 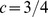 , então provamos que o tempo de descoberta esperado pela deriva aleatória é exponencial no comprimento da sequência
, então provamos que o tempo de descoberta esperado pela deriva aleatória é exponencial no comprimento da sequência 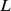 (veja a Figura 2 ). Esse resultado vale para qualquer tamanho de população,
(veja a Figura 2 ). Esse resultado vale para qualquer tamanho de população, 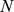 , desde que
, desde que 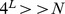 , o que certamente é o caso para valores realistas de
, o que certamente é o caso para valores realistas de 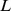 e
e 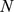 . No Texto S1, também apresentamos um resultado mais geral, onde junto com um único pico amplo, em vez de uma paisagem plana fora do pico, consideramos uma paisagem de aptidão multiplicativa e estabelecemos um resultado de dicotomia nítida que generaliza o Teorema 1 (veja o Corolário S2 no Texto S1 ).
. No Texto S1, também apresentamos um resultado mais geral, onde junto com um único pico amplo, em vez de uma paisagem plana fora do pico, consideramos uma paisagem de aptidão multiplicativa e estabelecemos um resultado de dicotomia nítida que generaliza o Teorema 1 (veja o Corolário S2 no Texto S1 ).
Figura 2. Pico amplo com diferentes paisagens de aptidão.

Para o pico amplo, há uma sequência específica, e todas as sequências que estão dentro da distância de Hamming 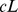 fazem parte do conjunto alvo. O cenário de aptidão é plano fora do pico amplo. (A) Se a largura do pico amplo for
fazem parte do conjunto alvo. O cenário de aptidão é plano fora do pico amplo. (A) Se a largura do pico amplo for 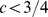 , então o tempo de descoberta esperado é exponencial em comprimento de sequência,
, então o tempo de descoberta esperado é exponencial em comprimento de sequência, 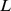 . (B) Se a largura do pico amplo for
. (B) Se a largura do pico amplo for 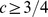 , então o tempo de descoberta esperado é polinomial em comprimento de sequência,
, então o tempo de descoberta esperado é polinomial em comprimento de sequência, 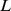 . (C) Cálculos numéricos para cenários de aptidão de pico amplo. Observamos o tempo de descoberta esperado exponencial para
. (C) Cálculos numéricos para cenários de aptidão de pico amplo. Observamos o tempo de descoberta esperado exponencial para 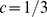 e
e 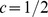 , enquanto o tempo de descoberta esperado polinomial para
, enquanto o tempo de descoberta esperado polinomial para 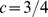 .
.
Observação 1
Destacamos dois aspectos importantes dos nossos resultados.
Primeiro, quando estabelecemos limites inferiores exponenciais para o tempo de descoberta esperado, esses limites inferiores são mantidos mesmo que a sequência inicial esteja a apenas alguns passos do conjunto alvo.
Em segundo lugar, apresentamos resultados de dicotomia forte e derivamos matematicamente a forma mais precisa e forte da condição de contorno.
Vamos agora dar um exemplo numérico para demonstrar que o tempo exponencial é intratável. A vida bacteriana na Terra existe há pelo menos 3,5 bilhões de anos, o que corresponde a 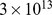 horas. Assumindo uma divisão celular bacteriana rápida de 20–30 minutos em média, temos no máximo
horas. Assumindo uma divisão celular bacteriana rápida de 20–30 minutos em média, temos no máximo 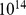 gerações. O tempo de descoberta esperado para uma sequência de comprimento
gerações. O tempo de descoberta esperado para uma sequência de comprimento 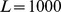 com um pico amplo muito grande de
com um pico amplo muito grande de 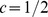 é de aproximadamente
é de aproximadamente 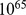 gerações; veja a Tabela 1 .
gerações; veja a Tabela 1 .
Tabela 1. Dados numéricos para tempo de descoberta em paisagens de aptidão planas.
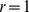
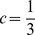
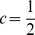
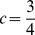
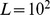
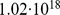
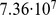
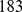
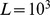
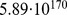
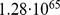
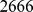
Dados numéricos para o tempo de descoberta de picos amplos com largura 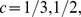 e
e 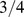 embutidos em paisagens de aptidão planas. Primeiro, o tempo de descoberta é computado para valores pequenos de
embutidos em paisagens de aptidão planas. Primeiro, o tempo de descoberta é computado para valores pequenos de 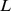 conforme mostrado na Figura 2(C) . Então, o crescimento exponencial é extrapolado para
conforme mostrado na Figura 2(C) . Então, o crescimento exponencial é extrapolado para 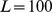 e
e 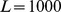 , respectivamente. Mostramos os tempos de descoberta para
, respectivamente. Mostramos os tempos de descoberta para 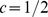 , e
, e 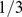 . Para
. Para 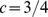 os valores são polinomiais em
os valores são polinomiais em 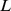 .
.
Se processos evolutivos individuais não conseguem encontrar alvos em tempo polinomial, então talvez o sucesso da evolução seja baseado no fato de que muitas populações estão buscando independentemente e em paralelo por uma adaptação particular. Nós provamos que múltiplas buscas paralelas independentes não são a solução do problema, se a sequência inicial estiver longe do centro alvo. Formalmente, mostramos o seguinte resultado.
Teorema 2
Em todos os casos em que o limite inferior do tempo de descoberta esperado é exponencial, para todos os polinômios , e , para qualquer sequência inicial com distância de Hamming de pelo menos a partir do centro do alvo, a probabilidade de qualquer uma das múltiplas pesquisas independentes atingir o conjunto alvo dentro de etapas é no máximo .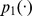
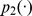
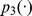
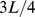
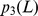
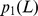
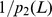
Se um processo evolutivo leva tempo exponencial, então polinomialmente muitas buscas independentes não encontram o alvo em tempo polinomial com probabilidade razoável (para detalhes veja Teorema S5 no Texto S1 ). Também mostramos um cálculo informal e aproximado da probabilidade de sucesso para 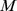 buscas independentes, como segue: se o tempo de descoberta esperado for exponencial (digamos,
buscas independentes, como segue: se o tempo de descoberta esperado for exponencial (digamos, 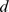 ), então a probabilidade de que todas
), então a probabilidade de que todas 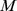 as buscas independentes falhem até
as buscas independentes falhem até 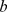 os passos é pelo menos
os passos é pelo menos 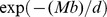 (ou seja, a probabilidade de sucesso dentro
(ou seja, a probabilidade de sucesso dentro 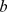 dos passos de qualquer uma das buscas é no máximo
dos passos de qualquer uma das buscas é no máximo 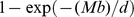 ), quando a sequência inicial está longe do centro alvo. Nesse caso, pode-se esgotar rapidamente os recursos físicos de um planeta inteiro. O número estimado de células bacterianas [36] na Terra é de cerca de
), quando a sequência inicial está longe do centro alvo. Nesse caso, pode-se esgotar rapidamente os recursos físicos de um planeta inteiro. O número estimado de células bacterianas [36] na Terra é de cerca de 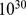 . Para dar um exemplo específico, vamos supor que existam
. Para dar um exemplo específico, vamos supor que existam 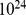 buscas independentes, cada uma com tamanho populacional
buscas independentes, cada uma com tamanho populacional 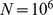 . A probabilidade de que pelo menos uma dessas buscas independentes tenha sucesso dentro de
. A probabilidade de que pelo menos uma dessas buscas independentes tenha sucesso dentro de 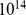 gerações para comprimento de sequência
gerações para comprimento de sequência 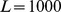 e pico amplo de
e pico amplo de 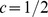 é menor que
é menor que 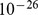 .
.
Em nosso modelo básico, mutantes individuais são avaliados um de cada vez. A situação de muitas linhagens mutantes evoluindo em paralelo é semelhante às buscas múltiplas descritas acima. Como mostramos que sempre que uma única busca leva tempo exponencial, buscas múltiplas independentes não levam a soluções de tempo polinomial, nossos resultados implicam intratabilidade para este caso também.
Agora exploramos o caso de múltiplos picos amplos que são distribuídos uniformemente e aleatoriamente. Considere que há  centros alvo. Ao redor de cada centro alvo há um gradiente de seleção que se estende até uma distância
centros alvo. Ao redor de cada centro alvo há um gradiente de seleção que se estende até uma distância 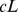 . Formalmente, podemos considerar qualquer função de aptidão
. Formalmente, podemos considerar qualquer função de aptidão 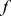 que atribui aptidão zero a uma sequência cuja distância de Hamming excede
que atribui aptidão zero a uma sequência cuja distância de Hamming excede 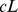 de todos os centros alvo, que em particular é subsumida ao considerar os múltiplos picos amplos onde ao redor de cada centro consideramos um pico amplo do conjunto alvo com largura de pico
de todos os centros alvo, que em particular é subsumida ao considerar os múltiplos picos amplos onde ao redor de cada centro consideramos um pico amplo do conjunto alvo com largura de pico  . Estabelecemos o seguinte resultado:
. Estabelecemos o seguinte resultado:
Teorema 3
Considere uma única busca sob o amplo panorama de aptidão de pico múltiplo de centros alvo escolhidos uniformemente ao acaso, com largura de pico no máximo para cada centro e . Então, com alta probabilidade, o tempo de descoberta esperado do conjunto alvo é pelo menos .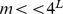

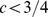
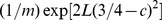
Se a função 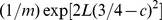 é exponencial ou não em
é exponencial ou não em 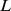 depende de como
depende de como  as mudanças com
as mudanças com 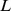 . Mas mesmo se assumirmos exponencialmente muitos centros de pico amplos,
. Mas mesmo se assumirmos exponencialmente muitos centros de pico amplos,  , com largura de pico
, com largura de pico 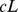 onde
onde 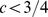 , não precisamos obter tempo polinomial ( Figura 3 e Teorema S6 no Texto S1 ).
, não precisamos obter tempo polinomial ( Figura 3 e Teorema S6 no Texto S1 ).
Figura 3. A busca por alvos distribuídos aleatoriamente e uniformemente no espaço de sequência.

(A) O conjunto alvo consiste em  sequências aleatórias; cada uma delas é cercada por um pico amplo de largura até
sequências aleatórias; cada uma delas é cercada por um pico amplo de largura até 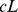 . A figura mostra uma ilustração pictórica onde o
. A figura mostra uma ilustração pictórica onde o 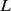 espaço de sequência -dimensional é projetado em duas dimensões. De uma sequência inicial escolhida aleatoriamente fora do conjunto alvo, o tempo de descoberta esperado é de pelo menos
espaço de sequência -dimensional é projetado em duas dimensões. De uma sequência inicial escolhida aleatoriamente fora do conjunto alvo, o tempo de descoberta esperado é de pelo menos 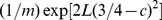 , que pode ser exponencial em
, que pode ser exponencial em 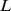 . (B) Simulações de computador mostrando o tempo médio de descoberta de alvos
. (B) Simulações de computador mostrando o tempo médio de descoberta de alvos 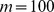 ,
, 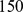 , e , com . Observamos dependência exponencial em . O tempo de descoberta é calculado em média em 200 execuções. (C) Probabilidade de sucesso estimada como a fração das 200 pesquisas que conseguem encontrar uma das sequências alvo dentro de gerações. A probabilidade de sucesso cai exponencialmente com . (D) Probabilidade de sucesso como uma função do tempo para e . (E) Tempo de descoberta para um grande número de sequências alvo geradas aleatoriamente. Ambas as sequências ou foram geradas. Para e o conjunto alvo consiste em bolas de distância de Hamming e (respectivamente) ao redor de cada sequência. A figura mostra o tempo médio de descoberta de 100 execuções. Como esperado, observamos que o tempo de descoberta cresce exponencialmente com o comprimento da sequência, .
, e , com . Observamos dependência exponencial em . O tempo de descoberta é calculado em média em 200 execuções. (C) Probabilidade de sucesso estimada como a fração das 200 pesquisas que conseguem encontrar uma das sequências alvo dentro de gerações. A probabilidade de sucesso cai exponencialmente com . (D) Probabilidade de sucesso como uma função do tempo para e . (E) Tempo de descoberta para um grande número de sequências alvo geradas aleatoriamente. Ambas as sequências ou foram geradas. Para e o conjunto alvo consiste em bolas de distância de Hamming e (respectivamente) ao redor de cada sequência. A figura mostra o tempo médio de descoberta de 100 execuções. Como esperado, observamos que o tempo de descoberta cresce exponencialmente com o comprimento da sequência, .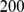
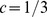
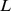
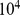
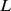
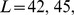
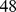
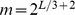
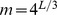
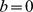
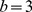


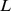
Sabe-se que a recombinação pode acelerar a evolução em certas paisagens de aptidão [28] , [37] – [39] , e a recombinação também pode desacelerar a evolução em outras paisagens de aptidão [40] . A recombinação, no entanto, reduz o tempo de descoberta apenas por um fator linear no comprimento da sequência [28] , [37] , [ 38] , [41] , [42] . Uma melhoria de fator linear ou mesmo polinomial sobre uma função exponencial não converte a função exponencial em uma função polinomial. Portanto, a recombinação pode fazer uma diferença significativa apenas se o processo evolutivo subjacente sem recombinação já operar em tempo polinomial.
Quais são então os problemas adaptativos que podem ser resolvidos pela evolução em tempo polinomial? Propomos um “processo de regeneração”. A ideia básica é que a evolução pode resolver um novo problema de forma eficiente, se já tiver resolvido um problema semelhante. Suponha que a duplicação genética ou o rearranjo do genoma podem dar origem a sequências iniciais que estão, no máximo, 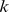 a mutações pontuais de distância do conjunto alvo, onde
a mutações pontuais de distância do conjunto alvo, onde 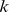 é um número que é independente de
é um número que é independente de 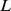 . É importante que as sequências iniciais possam ser regeneradas repetidamente. Provamos que
. É importante que as sequências iniciais possam ser regeneradas repetidamente. Provamos que 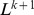 muitas buscas são suficientes para encontrar o alvo em tempo polinomial com alta probabilidade (veja a Figura 4 e a Seção 10 no Texto S1 ). O limite superior,
muitas buscas são suficientes para encontrar o alvo em tempo polinomial com alta probabilidade (veja a Figura 4 e a Seção 10 no Texto S1 ). O limite superior, 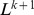 , vale mesmo para deriva neutra (sem seleção). Observe que, neste caso, o tempo de descoberta esperado para qualquer busca única ainda é exponencial. Portanto, a maioria das
, vale mesmo para deriva neutra (sem seleção). Observe que, neste caso, o tempo de descoberta esperado para qualquer busca única ainda é exponencial. Portanto, a maioria das 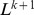 buscas não tem sucesso em tempo polinomial; no entanto, com alta probabilidade, uma das buscas tem sucesso em tempo polinomial. Existem dois aspectos principais no “processo de regeneração”: (a) a sequência inicial está a apenas um pequeno número de passos de distância do alvo; e (b) a sequência inicial pode ser gerada repetidamente. Este processo permite que a evolução supere a barreira exponencial. O limite superior,
buscas não tem sucesso em tempo polinomial; no entanto, com alta probabilidade, uma das buscas tem sucesso em tempo polinomial. Existem dois aspectos principais no “processo de regeneração”: (a) a sequência inicial está a apenas um pequeno número de passos de distância do alvo; e (b) a sequência inicial pode ser gerada repetidamente. Este processo permite que a evolução supere a barreira exponencial. O limite superior, 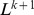 , pode possivelmente ser ainda mais reduzido, se a seleção e/ou recombinação forem incluídas.
, pode possivelmente ser ainda mais reduzido, se a seleção e/ou recombinação forem incluídas.
Figura 4. Processo de regeneração.

A duplicação de genes (ou possivelmente algum outro processo) gera um fluxo constante de sequências iniciais que estão a um número constante 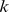 de mutações de distância do alvo. Muitas buscas se afastam do alvo, mas algumas terão sucesso em polinomialmente muitas etapas. Provamos que
de mutações de distância do alvo. Muitas buscas se afastam do alvo, mas algumas terão sucesso em polinomialmente muitas etapas. Provamos que 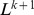 as buscas garantem que com alta probabilidade algumas buscas tenham sucesso em polinomialmente muitas etapas.
as buscas garantem que com alta probabilidade algumas buscas tenham sucesso em polinomialmente muitas etapas.
Discussão
O processo de regeneração formaliza o papel de várias ideias existentes. Primeiro, ele se vincula à proposta de que duplicações genéticas e rearranjos genômicos são eventos importantes que levam ao surgimento de novos genes [43] . Segundo, a evolução pode ser vista como um consertador brincando com pequenas modificações de sequências existentes em vez de criar sequências inteiramente novas [44] . Terceiro, o processo está relacionado à sugestão de Gillespie [29] de que a sequência inicial para uma busca evolutiva deve ter alta aptidão. Em nossa teoria, a proximidade no valor de aptidão é substituída pela proximidade no espaço de sequência. No entanto, nossos resultados mostram que a proximidade por si só é insuficiente para quebrar a barreira exponencial e, somente quando combinada com o processo de regeneração, produz tempo de descoberta polinomial com alta probabilidade. Nosso processo também pode explicar o surgimento de genes órfãos decorrentes de regiões não codificadoras [45] . A Seção 12 do Texto S1 discute a conexão de nossa abordagem com os resultados existentes.
Há um outro cenário que deve ser mencionado. É possível que certas funções biológicas sejam hiperabundantes no espaço de sequência [21] e que um processo que gere um grande número de sequências aleatórias encontre a função com alta probabilidade. Por exemplo, Bartel & Szostak [46] isolaram uma nova ribozima de um conjunto de 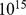 sequências aleatórias de comprimento
sequências aleatórias de comprimento 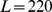 . Embora tal processo seja concebível para um comprimento de sequência efetivo pequeno, ele não pode representar uma solução geral para um comprimento de sequência grande
. Embora tal processo seja concebível para um comprimento de sequência efetivo pequeno, ele não pode representar uma solução geral para um comprimento de sequência grande 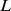 .
.
Nossa teoria tem implicações empíricas claras. O processo de regeneração pode ser testado em sistemas de evolução in vitro [47] . Uma sequência inicial pode ser gerada pela introdução de 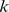 mutações pontuais em uma sequência de codificação de proteína conhecida de comprimento
mutações pontuais em uma sequência de codificação de proteína conhecida de comprimento 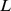 . Se essas mutações pontuais destruírem a função da proteína, então o tempo de descoberta esperado de qualquer tentativa de encontrar a sequência original deve ser exponencial em
. Se essas mutações pontuais destruírem a função da proteína, então o tempo de descoberta esperado de qualquer tentativa de encontrar a sequência original deve ser exponencial em 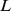 . Mas apenas muitas pesquisas polinomiais em
. Mas apenas muitas pesquisas polinomiais em 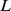 são necessárias para encontrar o alvo com alta probabilidade em muitas etapas polinomiais. A mesma configuração pode ser usada para explorar se a função biológica pode ser encontrada em outro lugar no espaço de sequência: a trajetória evolutiva começando com a sequência inicial poderia descobrir novas soluções. Nossa teoria também destaca o quão importante é explorar a distribuição de funções biológicas no espaço de sequência tanto para RNA [20] , [21] , [35] , [46] quanto no universo de proteínas [48] .
são necessárias para encontrar o alvo com alta probabilidade em muitas etapas polinomiais. A mesma configuração pode ser usada para explorar se a função biológica pode ser encontrada em outro lugar no espaço de sequência: a trajetória evolutiva começando com a sequência inicial poderia descobrir novas soluções. Nossa teoria também destaca o quão importante é explorar a distribuição de funções biológicas no espaço de sequência tanto para RNA [20] , [21] , [35] , [46] quanto no universo de proteínas [48] .
Em resumo, desenvolvemos uma teoria que nos permite estimar escalas de tempo de trajetórias evolutivas. Mostramos que vários processos naturais de evolução levam tempo exponencial como função do comprimento da sequência, 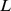 . Em alguns casos, estabelecemos resultados de dicotomia forte para condições de contorno precisas. Propusemos um mecanismo que permite a evolução em escalas de tempo polinomiais. Algumas direções interessantes de trabalho futuro são as seguintes: (1) Considere várias formas de paisagens de aptidão robustas e estude aproximações mais refinadas em comparação com as que consideramos; e então estime o tempo de descoberta esperado para as aproximações refinadas. (2) Enquanto neste artigo caracterizamos a diferença entre exponencial e polinomial para o tempo de descoberta esperado, análises mais refinadas (como eficiência para tempo polinomial, como tempo cúbico vs. quadrático) para paisagens de aptidão específicas usando mecanismos como recombinação é outro problema interessante.
. Em alguns casos, estabelecemos resultados de dicotomia forte para condições de contorno precisas. Propusemos um mecanismo que permite a evolução em escalas de tempo polinomiais. Algumas direções interessantes de trabalho futuro são as seguintes: (1) Considere várias formas de paisagens de aptidão robustas e estude aproximações mais refinadas em comparação com as que consideramos; e então estime o tempo de descoberta esperado para as aproximações refinadas. (2) Enquanto neste artigo caracterizamos a diferença entre exponencial e polinomial para o tempo de descoberta esperado, análises mais refinadas (como eficiência para tempo polinomial, como tempo cúbico vs. quadrático) para paisagens de aptidão específicas usando mecanismos como recombinação é outro problema interessante.
Materiais e métodos
Nossos resultados são baseados em uma análise matemática dos processos estocásticos subjacentes. Para cadeias de Markov na grade unidimensional, descrevemos relações de recorrência para o tempo de acerto esperado e apresentamos limites inferior e superior no tempo de acerto esperado usando análise combinatória (veja o Texto S1 para detalhes). Agora apresentamos os argumentos intuitivos básicos dos principais resultados.
Cadeia de Markov na grade unidimensional
Para um único pico amplo, devido à simetria, podemos interpretar a caminhada aleatória evolucionária como uma cadeia de Markov na grade unidimensional. Uma sequência do tipo  está
está  a passos de distância do alvo, onde
a passos de distância do alvo, onde  é a distância de Hamming entre esta sequência e o alvo. A probabilidade de que uma
é a distância de Hamming entre esta sequência e o alvo. A probabilidade de que uma  sequência de tipo sofra mutação para uma
sequência de tipo sofra mutação para uma  sequência de tipo é dada por
sequência de tipo é dada por 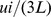 . O processo estocástico da caminhada aleatória evolucionária é uma cadeia de Markov na grade unidimensional
. O processo estocástico da caminhada aleatória evolucionária é uma cadeia de Markov na grade unidimensional  .
.
A relação de recorrência básica
Considere uma cadeia de Markov na grade unidimensional, e deixe 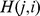 denotar o tempo de acerto esperado de
denotar o tempo de acerto esperado de  a
a 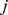 . A relação de recorrência geral para o tempo de acerto esperado é a seguinte:
. A relação de recorrência geral para o tempo de acerto esperado é a seguinte:
| (1) |
para 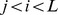 , com condição de contorno
, com condição de contorno 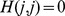 . A interpretação é a seguinte. Dado o estado atual
. A interpretação é a seguinte. Dado o estado atual  , se
, se 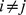 , pelo menos uma transição será feita para um estado vizinho
, pelo menos uma transição será feita para um estado vizinho 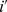 , com probabilidade
, com probabilidade 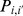 , do qual o tempo de acerto é
, do qual o tempo de acerto é 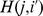 .
.
Intuição por trás do Teorema 1
O Teorema 1 é derivado pela obtenção de limites precisos para a relação de recorrência do tempo de acerto ( Equação 1 ). Considere que 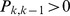 para todos
para todos 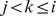 (ou seja, o progresso em direção ao estado
(ou seja, o progresso em direção ao estado 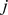 é sempre possível), pois de outra forma
é sempre possível), pois de outra forma 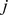 nunca é alcançado a partir de
nunca é alcançado a partir de  . Mostramos (veja Lema 2 no Texto S1 ) que podemos escrever
. Mostramos (veja Lema 2 no Texto S1 ) que podemos escrever 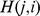 como uma soma,
como uma soma, 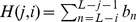 , onde
, onde 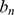 é a sequência definida como:
é a sequência definida como:
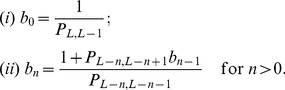 | (2) |
A intuição básica obtida da Equação 2 é a seguinte: (i) Se 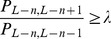 , para alguma constante
, para alguma constante 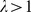 , então a sequência
, então a sequência  cresce pelo menos tão rápido quanto uma série geométrica com fator
cresce pelo menos tão rápido quanto uma série geométrica com fator  . (ii) Por outro lado, se
. (ii) Por outro lado, se 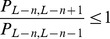 e
e 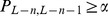 para alguma constante
para alguma constante 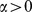 , então a sequência
, então a sequência 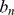 cresce no máximo tão rápido quanto uma série aritmética com diferença
cresce no máximo tão rápido quanto uma série aritmética com diferença 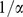 . Da análise de caso acima, o resultado para o Teorema 1 é obtido da seguinte forma: Se
. Da análise de caso acima, o resultado para o Teorema 1 é obtido da seguinte forma: Se 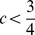 , então para todo
, então para todo 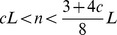 , temos
, temos 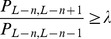 para algum
para algum 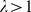 , e, portanto, a sequência
, e, portanto, a sequência 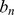 cresce geometricamente para um comprimento linear em
cresce geometricamente para um comprimento linear em 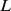 . Então,
. Então, 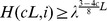 para todos os estados
para todos os estados 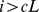 (ou seja, para todas as sequências fora do conjunto alvo). Isso corresponde ao caso 1 do Teorema 1. Por outro lado, se
(ou seja, para todas as sequências fora do conjunto alvo). Isso corresponde ao caso 1 do Teorema 1. Por outro lado, se 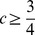 , então é
, então é 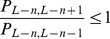 , e o caso 2 do Teorema 1 é derivado (para detalhes, veja Corolário 2 no Texto S1 ).
, e o caso 2 do Teorema 1 é derivado (para detalhes, veja Corolário 2 no Texto S1 ).
Intuição por trás do Teorema 2
A intuição básica para o resultado é a seguinte: considere uma única busca para a qual o tempo de acerto esperado é exponencial. Então, para a busca única, a probabilidade de sucesso em muitos passos polinomiais é desprezível (pois, de outra forma, a expectativa não teria sido exponencial). No caso de buscas independentes, a independência garante que a probabilidade de que todas as buscas falhem é o produto das probabilidades de que cada busca individual falhe. Usando os argumentos acima, estabelecemos o Teorema 2 (para detalhes, veja a Seção 8 no Texto S1 ).
Intuição por trás do Teorema 3
Para este resultado, é conveniente primeiro visualizar a caminhada evolutiva ocorrendo no espaço de sequência de todas as sequências de comprimento 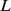 , sob nenhuma seleção. Cada sequência tem
, sob nenhuma seleção. Cada sequência tem 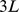 vizinhos e, considerando que uma mutação pontual acontece, a probabilidade de transição para cada um deles é
vizinhos e, considerando que uma mutação pontual acontece, a probabilidade de transição para cada um deles é 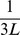 . A cadeia de Markov subjacente devido à simetria tem tempo de mistura rápido, ou seja, o número de etapas para convergir para a distribuição estacionária (o tempo de mistura) é
. A cadeia de Markov subjacente devido à simetria tem tempo de mistura rápido, ou seja, o número de etapas para convergir para a distribuição estacionária (o tempo de mistura) é 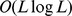 . Novamente por simetria, a distribuição estacionária é a distribuição uniforme. Se
. Novamente por simetria, a distribuição estacionária é a distribuição uniforme. Se 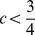 , então do Teorema 1 obtemos que o tempo esperado para atingir um único pico amplo é exponencial. Por limite de união, se
, então do Teorema 1 obtemos que o tempo esperado para atingir um único pico amplo é exponencial. Por limite de união, se 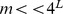 , a probabilidade de atingir qualquer um dos
, a probabilidade de atingir qualquer um dos  picos amplos dentro
picos amplos dentro 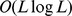 das etapas é desprezível. Como após as primeiras
das etapas é desprezível. Como após as primeiras 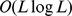 etapas a cadeia de Markov converge para a distribuição estacionária, então cada etapa do processo pode ser interpretada como seleção de sequências uniformemente aleatórias entre todas as sequências. Usando a desigualdade de Hoeffding, mostramos que com alta probabilidade, na expectativa,
etapas a cadeia de Markov converge para a distribuição estacionária, então cada etapa do processo pode ser interpretada como seleção de sequências uniformemente aleatórias entre todas as sequências. Usando a desigualdade de Hoeffding, mostramos que com alta probabilidade, na expectativa, 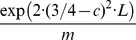 tais etapas são necessárias antes que uma sequência seja encontrada que pertença ao conjunto alvo. Assim obtemos o resultado do Teorema 3 (para detalhes veja Seção 9 no Texto S1 ).
tais etapas são necessárias antes que uma sequência seja encontrada que pertença ao conjunto alvo. Assim obtemos o resultado do Teorema 3 (para detalhes veja Seção 9 no Texto S1 ).
Comentário sobre técnicas
Um aspecto importante do nosso trabalho é que estabelecemos nossos resultados usando técnicas elementares para análise de cadeias de Markov. O uso de maquinário matemático mais avançado, como martingales [49] ou análise de deriva [50] , [51] , pode possivelmente ser usado para derivar resultados mais refinados. Embora neste trabalho nosso objetivo seja distinguir entre tempo exponencial e polinomial, se as técnicas de [49] – [51] podem levar a uma caracterização mais refinada dentro do tempo polinomial é uma direção interessante para trabalhos futuros.
Informações de suporte
Provas detalhadas para “A escala de tempo da inovação evolucionária”.
(PDF)
Agradecimentos
Agradecemos a Nick Barton e Daniel Weissman pelas discussões úteis e por nos indicarem literatura relevante.
Declaração de financiamento
Austrian Science Fund (FWF) Grant No P23499-N23, FWF NFN Grant No S11407-N23 (RiSE), ERC Start Grant (279307: Graph Games) e prêmio Microsoft Faculty Fellows. O apoio da John Templeton Foundation é reconhecido com gratidão. Os financiadores não tiveram nenhum papel no desenho do estudo, coleta e análise de dados, decisão de publicação ou preparação do manuscrito.
Referências
- 1. Allwood AC, Grotzinger JP, Knoll AH, Burch IW, Anderson MS, et al. (2009) Controles no desenvolvimento e diversidade de estromatólitos arqueanos iniciais. Proc Natl Acad Sci USA 106: 9548–9555. [ DOI ] [ Artigo gratuito PMC ] [ PubMed ] [ Google Scholar ]
- 2. Schopf JW (agosto de 2006) O primeiro bilhão de anos: Quando surgiu a vida? Elementos 2: 229–233. [ Google Scholar ]
- 3. Kimura M (1968) Taxa evolutiva no nível molecular. Natureza 217: 624–626. [ DOI ] [ PubMed ] [ Google Scholar ]
- 4. Ewens WJ (1967) A probabilidade de sobrevivência de um novo mutante em um ambiente flutuante. Heredity 22: 438–443. [ DOI ] [ PubMed ] [ Google Scholar ]
- 5. Barton NH (1995) Ligação e os limites da seleção natural. Genética 140: 821–41. [ DOI ] [ PMC artigo gratuito ] [ PubMed ] [ Google Scholar ]
- 6. Campos PR (2004) Fixação de mutações benéficas na presença de interações epistáticas. Bull Math Biol 66: 473–486. [ DOI ] [ PubMed ] [ Google Scholar ]
- 7. Antal T, Scheuring I (2006) Fixação de estratégias para um jogo evolucionário em populações finitas. Bull Math Biol 68: 1923–1944. [ DOI ] [ PubMed ] [ Google Scholar ]
- 8. Whitlock MC (2003) Probabilidade e tempo de fixação em populações subdivididas. Genética 164: 767–779. [ DOI ] [ PMC artigo gratuito ] [ PubMed ] [ Google Scholar ]
- 9.Altrock PM, Traulsen A (2009) Tempos de fixação em jogos evolucionários sob seleção fraca. New J Phys 11 . doi:10.1088/1367-2630/11/1/013012 [ Google Scholar ]
- 10. Kimura M, Ohta T (1969) Número médio de gerações até a fixação de um gene mutante em uma população finita. Genetics 61: 763–771. [ DOI ] [ PMC artigo gratuito ] [ PubMed ] [ Google Scholar ]
- 11. Johnson T, Gerrish P (2002) A probabilidade de fixação de um alelo benéfico em uma população que se divide por fissão binária. Genetica 115: 283–287. [ DOI ] [ PubMed ] [ Google Scholar ]
- 12. Orr HA (2000) A taxa de adaptação em assexuais. Genética 155: 961–968. [ DOI ] [ PMC artigo gratuito ] [ PubMed ] [ Google Scholar ]
- 13. Wilke CO (2004) A velocidade de adaptação em grandes populações assexuadas. Genética 167: 2045–2053. [ DOI ] [ PMC artigo gratuito ] [ PubMed ] [ Google Scholar ]
- 14. Desai MM, Fisher DS, Murray AW (2007) A velocidade da evolução e manutenção da variação em populações assexuadas. Curr Biol 17: 385–394. [ DOI ] [ PMC artigo gratuito ] [ PubMed ] [ Google Scholar ]
- 15. Ohta T (1972) Tamanho populacional e taxa de evolução. J Mol Evol 1: 305–314. [ PubMed ] [ Google Scholar ]
- 16.Papadimitriou C (1994) Complexidade computacional. Addison-Wesley.
- 17.Cormen T, Leiserson C, Rivest R, Stein C (2009) Introdução aos Algoritmos. Imprensa do MIT.
- 18. Valiant LG (2009) Evolutividade. J ACM 56: 3:1–3:21. [ Google Scholar ]
- 19. Maynard Smith J (1970) Seleção natural e o conceito de um espaço proteico. Nature 225: 563–564. [ DOI ] [ PubMed ] [ Google Scholar ]
- 20. Fontana W, Schuster P (1987) Um modelo computacional de otimização evolutiva. Biophys Chem 26: 123–147. [ DOI ] [ PubMed ] [ Google Scholar ]
- 21. Fontana W, Schuster P (1998) Continuidade na evolução: Sobre a natureza das transições. Science 280: 1451–1455. [ DOI ] [ PubMed ] [ Google Scholar ]
- 22. Eigen M, Mccaskill J, Schuster P (1988) Quase-espécies moleculares. J Phys Chem 92: 6881–6891. [ Google Scholar ]
- 23. Eigen M, Schuster P (1978) O hiperciclo. Naturwissenschaften 65: 7–41. [ DOI ] [ PubMed ] [ Google Scholar ]
- 24. Park SC, Simon D, Krug J (2010) A velocidade da evolução em grandes populações assexuadas. J Stat Phys 138: 381–410. [ Google Scholar ]
- 25. Derrida B, Peliti L (1991) Evolução em um cenário de aptidão física. Bull Math Biol 53: 355–382. [ Google Scholar ]
- 26. Stadler PF (2002) Paisagens de aptidão. Appl Math & Comput 117: 187–207. [ Google Scholar ]
- 27. Worden RP (1995) Um limite de velocidade para a evolução. J Theor Biol 176: 137–152. [ DOI ] [ PubMed ] [ Google Scholar ]
- 28.Crow JF, Kimura M (1965) Evolução em populações sexuais e assexuadas. Am Nat 99 : pp. 439–450. [ Google Scholar ]
- 29. Gillespie JH (1984) Evolução molecular sobre a paisagem mutacional. Evolution 38: 1116–1129. [ DOI ] [ PubMed ] [ Google Scholar ]
- 30. Kauffman S, Levin S (1987) Em direção a uma teoria geral de caminhadas adaptativas em paisagens acidentadas. Journal of Theoretical Biology 128: 11–45. [ DOI ] [ PubMed ] [ Google Scholar ]
- 31. Orr HA (2000) Um mínimo no número médio de passos dados em caminhadas adaptativas. Journal of Theoretical Biology 220: 241–247. [ DOI ] [ PubMed ] [ Google Scholar ]
- 32. Weinreich DM, Watson RA, Chao L (2005) Perspectiva: epistasia de sinais e restrição genética em trajetórias evolutivas. Evolution 59: 1165–1174. [ PubMed ] [ Google Scholar ]
- 33. Poelwijk FJ, Kiviet DJ, Weinreich DM, Tans SJ (2007) Paisagens de aptidão empírica revelam caminhos evolutivos acessíveis. Nature 445: 383–386. [ DOI ] [ PubMed ] [ Google Scholar ]
- 34. Woods RJ, Barrick JE, Cooper TF, Shrestha U, Kauth MR, et al. (2011) Seleção de segunda ordem para evolutividade em uma grande população de escherichia coli. Science 331: 1433–1436. [ DOI ] [ PMC artigo gratuito ] [ PubMed ] [ Google Scholar ]
- 35. Jimenez JI, Xulvi-Brunet R, Campbell GW, Turk-MacLeod R, Chen IA (2013) Cenário abrangente de aptidão experimental e rede evolutiva para pequeno rna. Proc Natl Acad Sci USA. 110(37): 14984–9. [ DOI ] [ Artigo gratuito PMC ] [ PubMed ] [ Google Scholar ]
- 36. Whitman WB, Coleman DC, Wiebe WJ (1998) Procariotos: A maioria invisível. Proc Natl Acad Sci USA 95: 6578–6583. [ DOI ] [ PMC artigo gratuito ] [ PubMed ] [ Google Scholar ]
- 37. Smith JM (1974) Recombinação e a taxa de evolução. Genética 78: 299–304. [ DOI ] [ PMC artigo gratuito ] [ PubMed ] [ Google Scholar ]
- 38.Crow JF, Kimura M (1970) Uma introdução à teoria da genética populacional. Burgess Publishing Company.
- 39. Park SC, Krug J (2013) Taxa de adaptação em sexuais e assexuais: Um modelo solucionável do efeito fishermuller. Genetics 195: 941–955. [ DOI ] [ PMC artigo gratuito ] [ PubMed ] [ Google Scholar ]
- 40. de Visser JAGM, Park S, Krug J (2009) Explorando o efeito do sexo em paisagens de aptidão empírica. The American Naturalist 174: S15–S30. [ DOI ] [ PubMed ] [ Google Scholar ]
- 41. Neher RA, Shraiman BI, Fisher DS (2010) Taxa de adaptação em grandes populações sexuais. Genética 184: 467–481. [ DOI ] [ PMC artigo gratuito ] [ PubMed ] [ Google Scholar ]
- 42. Weissman DB, Hallatschek O (2014) A taxa de adaptação em grandes populações sexuais com cromossomos lineares. Genética 196: 1167–1183. [ DOI ] [ PMC artigo gratuito ] [ PubMed ] [ Google Scholar ]
- 43.Ohno S (1970) Evolução por duplicação genética. Springer-Verlag.
- 44. Jacob F (1977) Evolução e ajustes. Science 196: 1161–1166. [ DOI ] [ PubMed ] [ Google Scholar ]
- 45. Tautz D, Domazet-Lošo T (2011) A origem evolutiva dos genes órfãos. Nat Rev Genet 12: 692–702. [ DOI ] [ PubMed ] [ Google Scholar ]
- 46. Bartel D, Szostak J (1993) Isolamento de novas ribozimas de um grande conjunto de sequências aleatórias. Science 261: 1411–1418. [ DOI ] [ PubMed ] [ Google Scholar ]
- 47. Leconte AM, Dickinson BC, Yang DD, Chen IA, Allen B, et al. (2013) Um modelo experimental baseado em população para evolução de proteínas: efeitos da taxa de mutação e rigor de seleção em resultados evolutivos. Bioquímica 52: 1490–1499. [ DOI ] [ Artigo gratuito PMC ] [ PubMed ] [ Google Scholar ]
- 48. Povolotskaya IS, Kondrashov FA (2010) Espaço de sequência e a expansão contínua do universo de proteínas. Nature 465: 922–926. [ DOI ] [ PubMed ] [ Google Scholar ]
- 49.Williams D (1991) Probabilidade com Martingales. Livros didáticos matemáticos de Cambridge. Cambridge University Press.
- 50.Hajek B (1982) Limites de tempo de acerto e tempo de ocupação implícitos pela análise de deriva com aplicações. Advances in Applied Probability 14 : pp. 502–525. [ Google Scholar ]
- 51.Lehre PK, Witt C (2013) Análise geral de deriva com limites de cauda. CoRR abs/1307.2559.
Dados associados
Esta seção coleta quaisquer citações de dados, declarações de disponibilidade de dados ou materiais suplementares incluídos neste artigo.
Materiais suplementares
Provas detalhadas para “A escala de tempo da inovação evolucionária”.
(PDF)
Arquivo adicional
https://pmc.ncbi.nlm.nih.gov/articles/instance/4161296/bin/pcbi.1003818.s001.pdf
https://translate.google.com/?hl=pt-PT&eotf=0&sl=en&tl=pt&op=translate
Tradução: Inglês - Português
2 -
Comentários
Postar um comentário